



Next: References
Up: Substitution matrices
Previous: Codon usage table
Contents
In our model, the probability that a nucleotide mutation occurs at the
DNA level and the probability that the mutation is accepted (i.e. is
functional) at the protein level are separated into the nucleotide and
amino acid matrices. In contrast, the widely used BLOSUM (Henikoff &
Henikoff 1992) and PAM (Dayhoff et al. 1978) matrices
incorporate both effects into one matrix. In the PAM matrices, the
small-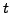 amino acid substitution frequencies are extrapolated to
larger . This is a serious short-coming since, in reality, at
small a mutating sequence is constrained to resemble the original
sequence at both the nucleotide and amino acid levels, whereas at
large a mutating sequence is only constrained to resemble the
original at the amino acid level. On the other hand, the BLOSUM
matrices are calculated, in effect, for a series of values:
BLOSUM100, BLOSUM95, ... BLOSUM35, with the lower indices
corresponding to more divergent sequences. By choosing a low-index
BLOSUM matrix (viz. BLOSUM40) as our default amino acid distance
matrix
amino acid substitution frequencies are extrapolated to
larger . This is a serious short-coming since, in reality, at
small a mutating sequence is constrained to resemble the original
sequence at both the nucleotide and amino acid levels, whereas at
large a mutating sequence is only constrained to resemble the
original at the amino acid level. On the other hand, the BLOSUM
matrices are calculated, in effect, for a series of values:
BLOSUM100, BLOSUM95, ... BLOSUM35, with the lower indices
corresponding to more divergent sequences. By choosing a low-index
BLOSUM matrix (viz. BLOSUM40) as our default amino acid distance
matrix 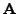 , we minimize the effect of the nucleotide mutation
constraint relative to the amino acid acceptability constraint.
, we minimize the effect of the nucleotide mutation
constraint relative to the amino acid acceptability constraint.
We use the scaled observed frequencies (Henikoff & Henikoff
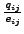 values) rather than log odds scores, and treat
values) rather than log odds scores, and treat
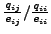 as the probability of
acceptance for the amino acid substitution
as the probability of
acceptance for the amino acid substitution
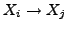 relative to
relative to
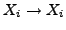 which is unity. The
which is unity. The 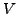 parameter
(
parameter
(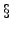 12.3) scales the off-diagonal terms of
relative to the diagonal terms, with the default value
12.3) scales the off-diagonal terms of
relative to the diagonal terms, with the default value 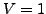 giving the original BLOSUM40 matrix. Stop codons are also
included, with the acceptabilities for mutations between stops and
non-stops set to zero.
giving the original BLOSUM40 matrix. Stop codons are also
included, with the acceptabilities for mutations between stops and
non-stops set to zero.
Next: References
Up: Substitution matrices
Previous: Codon usage table
Contents
aef
2007-12-10