HOME
Scan for Motifs is an approach to simplify the process of identifying a wide range of regulatory elements in RNA. It can also be used to search any nucleotide sequences. It is particularly useful for RNA-binding protein sites (RBPDB), Translational control elements (Transterm), targets of conserved miRNA families (TargetScan) as well as 6-8 base long miRNA seed sequence targets (mirBase) on alignments of 3' UTR sequences
The standalone webserver is here and is also part of the Transterm site.
Please cite: Biswas, A. and C. M. Brown (2014) Scan for Motifs: a webserver for the analysis of post-transcriptional regulatory elements in the 3' untranslated regions (3' UTRs) of mRNAs BMC Bioinformatics 15: 174.link
For enquiries please contact Ambarish Biswas or Dr. Chris Brown
Last update 26/4/2018
Example: >SEQUENCE_1 UGGCCACCACCUGGAAUUCAGAAUGGGGCGCCCAGAACGCACCAGGGCCUGGGGUUGGGAUUCCCGAGUGGGAGCCCUUGGGGCGCUGGGAAUGCGGAGGCCGGGGCUGCUUUGGCUCCUGUCAGAGCCCUUCGGCCAUCCCUGACCU AGAACCUGACGUGAGUGGACCCCAGACCUCCCGCUCUCCAGGUGUUUCCAGACUGUUCCCUGAGAGCGGAGCCCAGCCCCUGCCCCUCCCCACAGGACGCACUCCCUAUUUAUGUUUGCACUAGAGGUUAUUUAUUAUUUAUUUAUUA UUUAUUUAUUGACCAAUUAACUUAUUUAUUCGGGAGGUUGGGGUGUCCCAGGGGACCCAGCGUAGGGACAGCCUUGGCUCUGGCGUGUUUUCUGUGAAAACGGAGCCGAGCCGUGGGCUGCUCCCCCUUGGCCUCCUGGCCUCCGUGC CUCCCUUCGCUUAUGUUUUGAAGAAAUAUUUAUCUGAUCAAGUUGUCUGAAUAAUGCUGAUUUGGUGACAGGCUGUCGCUACAUCGCUGAACCUCUGCUCCCCAGGGGAGUUUUGUCUGUAACCGCCCUACUGGUCAGUGGCGAGAAA UAAAAUGUGCUUAGAAAAG >SEQUENCE_2 GGAGACUGGACAUUCAUCUUCACCUGGCUCAAAUCUUUUAGUAGCCACUCCUCCACACCCCCCUCCCCUAUUUAUUUCUGGUUUAGAAAGGGAAUUAGGGCCUCCGGGCCAGGCCCCAAGCUUGGAACUUUAAACAACAACACUUAAA ACCUAGGAUGUGAAGAUGUAUGGCCUGAACAAUGGGGCACUGGCUACCACAUAGAGUUCAGACUAGGGCUCCCAGAACUCACUGGGAGUCUGAAAUCUGGAUUCCUGAGUGCAGCCUAGGACACCUGGAAUGUGCAAGUCAGGGAAUC CUUGGUUCUGGUCAGAACAUCUCUUGAGAAGAUCUCACUUAGAACUUGACACAAGUGGACCUCAGGUCUCCCUUUCUUCAGAUGUCUCCAGACUCUCCUGAGAUGGAGAGCCCAGCCCCUCUUGUCUCCCACAGGGCCAGUUCUUUCU AUUUAUGUUUGCACUUGUGAUUAUUUAUUAUUUAUUUAUUAUUUAUUUAUUUACUGAUAAACCUAUUUAUUCAGGAGGUUAGUGUGUCCUGGGAGAGCCAGCAGAGGGGCUGCCUUGGCUUAGACAUGUUUUCUAUGAAAACGGAGCU GAACUAUAGGCUGUUCCCACCGGCCUCCAGGCCUCUGUGCCUUCUUUUGCAUAAUUUUGUUUUAAUUUAUCUGAUCAAGUUGUCUAAAUAAUGCUGAUUUGGUGACCUACUGUCGCUAUGUCGCUGAACCUCUGCUCCCCAGGGGAGU UGUCUUUGUAAUCGCCCAACUGAUCAGUGGCGAAAAAUAAAGUGUGCUUGGAAGUG >SEQUENCE_3 GGGGGAUGCAUGCAGAUCAUUCACCACCCAGCCCAUCGCCCUCCCUGUCCUGCCAUUCCCAUUGGGCCUCCUCGUCCCCGAAAGGAAGGGGGACGAGCCGGGUCUCUAAGUCAUCACCCCGAACAACAACACUUAGAACUUGAGAUGC AAGGAUGUGUGACUCAGACCAGACCGGGGGCACUGACCACCGCAGCCUGGAAUCCAAACUGGGGCUUGCAGAACCCACUGGGUCUCCAGAUGCAAAUGGGGACACCUGAAAUGUGGAGGUCUCCUUGAGCCUCCGGCUCACUUCCGAA GAUCUGAGGAGUCCUCACCCAGAACUUGGCAUGACCUCCGACCUCCCUUGCACCGAAAGUUUCUAGACCCUCCCCUAAGAGAUGAAGCCCCCCGCCACGUGGCACUAGUCAGCCCUCUAUUUAUGUUUGCACUGAGAAUUAUUUAUUA UUUAUUAUUUAUAUAUUUAUUUAUUUCCUGGUGAAUGUAUUUAUUCAGGAGGUCGGGGAACCUGGGGGAUCCAGUGUUGGGGGUUGCCUGAGCUCAGACAUGUUUUCUAUGAAAAUGGAGCUGGAAUGUAGGCUGCUCCCACCCCGCU UCCUGGCCUCCUUACCUCCCUGUGCUUGUGGAUUUAUCGAUCGAGUUGUCUGGAUAAUGCUGAUUUGGCGACAGACUGUUGCUAUCUCGCUGAACCUCUGCUCCCCAGGGGAGUUGUGCCUGUAAUCGUCCUACUGGUCAGUGGCGAA AAAUAAAGUUUGCUUAGAAAAG
Example:
Example:
CLUSTAL 2.1 multiple sequence alignment
SEQUENCE_1 ------------------------------------------------------------
SEQUENCE_2 GGAGACUGGACAUUCAUCUUCACCUGGCUCAAAUCUUUUAGUAGCCACUCCUCCACACCC
SEQUENCE_3 ------GGGGGAUGCAUGCAGAUCAUUCACCACCCAGCCCAUCGCCCUCCCUGUCCUGCC
SEQUENCE_1 ------------------------------------------------------------
SEQUENCE_2 CCCUCCCCUAUUUAUUUCUGGUUUAGAAAGGGAAUUAGGGCCUCCGGGCCAGGCCCCAAG
SEQUENCE_3 AUUCCCAUUGGGCCUCCUCGUCCCCGAAAGGAAGGGGGA-----CGAGCCGGGUCUCUAA
SEQUENCE_1 ------------------------------------------------------------
SEQUENCE_2 CUUGGAACUUUAAACAACAACACUUAAAACCUAGGAUGUGAAGAUGUAUGGCCUGAACAA
SEQUENCE_3 GUCAUCACCCCGAACAACAACACUUAGAACUUGAGAUGCAAGGAUGUGUGACUCAGACCA
SEQUENCE_1 ------------UGGCCACCACC---UGGAAUUCAGAAUGGGGCGCCCAGAACGCACCAG
SEQUENCE_2 U----GGGGCACUGGCUACCACA---UAGAGUUCAGACUAGGGCUCCCAGAACUCACUGG
SEQUENCE_3 GACCGGGGGCACUGACCACCGCAGCCUGGAAUCCAAACUGGGGCUUGCAGAACCCACUGG
** * *** * * ** * ** * * **** ****** *** *
SEQUENCE_1 G-GCCUGGGGUUGGGAUUCCCGAGUGGGAGCCCUUGGGGCGCUGGGAAUGCGGAGGCCGG
SEQUENCE_2 GAGUCUGAAAUCUGGAUUCCUGAGUGCAG---CCUAGGACACCUGGAAUGUGCAAGUCAG
SEQUENCE_3 G---------------UCUCCAGAUGCAA---AUGGGGACACCUGAAAUGUGGAGGUCUC
* * * ** ** * * * **** * * * *
SEQUENCE_1 G--GCUGCUUUGGCUCCUGUCAGAGC--CCUUCGGCCAUCCCUGACCUAGAACCUGACGU
SEQUENCE_2 G--GAAUCCUUGGUUCUGGUCAGAACAUCUCUUGAGAAGAUCUCACUUAGAACUUGACAC
SEQUENCE_3 CUUGAGCCUCCGGCUCACUUCCGAAGA---UCUGAGGAGUCCUCACCCAGAACUUGGCAU
* * ** ** ** ** * * ** ** ***** ** *
SEQUENCE_1 GAGUGGACCCCAGACCUCCCGCUCUCCAGGUGUUUCCAGACUGUUCCCUGAGA-GCGGAG
SEQUENCE_2 AAGUGGACCUCAGGUCUCCCUUUCUUCAGAUGUCUCCAGACUCU--CCUGAGAUGGAGAG
SEQUENCE_3 -----GACCUCCGACCUCCCUUGCACCGAAAGUUUCUAGACCCUCCCCUAAGAGAUGAAG
**** * * ***** * * ** ** **** * *** *** **
SEQUENCE_1 CCCAGCCCCUGCCCCUCCCCACAGGAC--GCACUCCCUAUUUAUGUUUGCACUAGAGGUU
SEQUENCE_2 CCCAGCCCCUCUUGUCUCCCACAGGGCCAGUUCUUUCUAUUUAUGUUUGCACUUGUGAUU
SEQUENCE_3 CCC----CCCGCCACGUGGCACUAGUC---AGCCCUCUAUUUAUGUUUGCACUGAGAAUU
*** ** *** * * * ***************** **
SEQUENCE_1 AUUUAUUAUUUAUU---UAU-UAUUUAUUUAUUGACCAAUUAACUUAUUUAUUCGGGAGG
SEQUENCE_2 AUUUAUUAUUUAUU---UAU-UAUUUAUUUAUUUACUGAUAAACCUAUUUAUUCAGGAGG
SEQUENCE_3 AUUUAUUAUUUAUUAUUUAUAUAUUUAUUUAUUUCCUGGUGAAUGUAUUUAUUCAGGAGG
************** *** ************ * * ** ********* *****
SEQUENCE_1 UUGGGGUGUCCCAGGGGACCCAGCGU-AGGGACAGCCUUGGCUCUGGCGUGUUUUCUGUG
SEQUENCE_2 UUAGUGUGUCCUGGGAGAGCCAGCAG-AGGGGCUGCCUUGGCUUAGACAUGUUUUCUAUG
SEQUENCE_3 UCGGGG-AACCUGGGGGAUCCAGUGUUGGGGGUUGCCUGAGCUCAGACAUGUUUUCUAUG
* * * ** ** ** **** *** **** *** * * ******** **
SEQUENCE_1 AAAACGGAGCCGAGCCGUGGGCUGCUCCCCCUUGGCCUCCUGGCCUCCGUGCCUCCCUUC
SEQUENCE_2 AAAACGGAGCUGAACUAUAGGCUGUUCCCACC-GGCCUCCAGGCCUCUGUGCCUUCUUUU
SEQUENCE_3 AAAAUGGAGCUGGAAUGUAGGCUGCUCCCACCCCGCUUCCUGGCCUCCUUACCUCCCUGU
**** ***** * * ***** **** * ** *** ****** * *** * *
SEQUENCE_1 GCUUAUGUUUUGAAGAAAUAUUUAUCUGAUCAAGUUGUCUGAAUAAUGCUGAUUUGGUGA
SEQUENCE_2 GCAUA-AUUUUGUUUUAA--UUUAUCUGAUCAAGUUGUCUAAAUAAUGCUGAUUUGGUGA
SEQUENCE_3 GCUUG---------UGGA--UUUAUC-GAUCGAGUUGUCUGGAUAAUGCUGAUUUGGCGA
** * * ****** **** ******** *************** **
SEQUENCE_1 CAGGCUGUCGCUACAUCGCUGAACCUCUGCUCCCCAGGGGAGUUUUGUCUGUAACCGCCC
SEQUENCE_2 CCUACUGUCGCUAUGUCGCUGAACCUCUGCUCCCCAGGGGAGUUGUCUUUGUAAUCGCCC
SEQUENCE_3 CAGACUGUUGCUAUCUCGCUGAACCUCUGCUCCCCAGGGGAGUUGUGCCUGUAAUCGUCC
* **** **** ***************************** * ***** ** **
SEQUENCE_1 UACUGGUCAGUGGCGAGAAAUAAAAUGUGCUUAGAAAAG
SEQUENCE_2 AACUGAUCAGUGGCGAAAAAUAAAGUGUGCUUGGAAGUG
SEQUENCE_3 UACUGGUCAGUGGCGAAAAAUAAAGUUUGCUUAGAAAAG
**** ********** ******* * ***** *** *
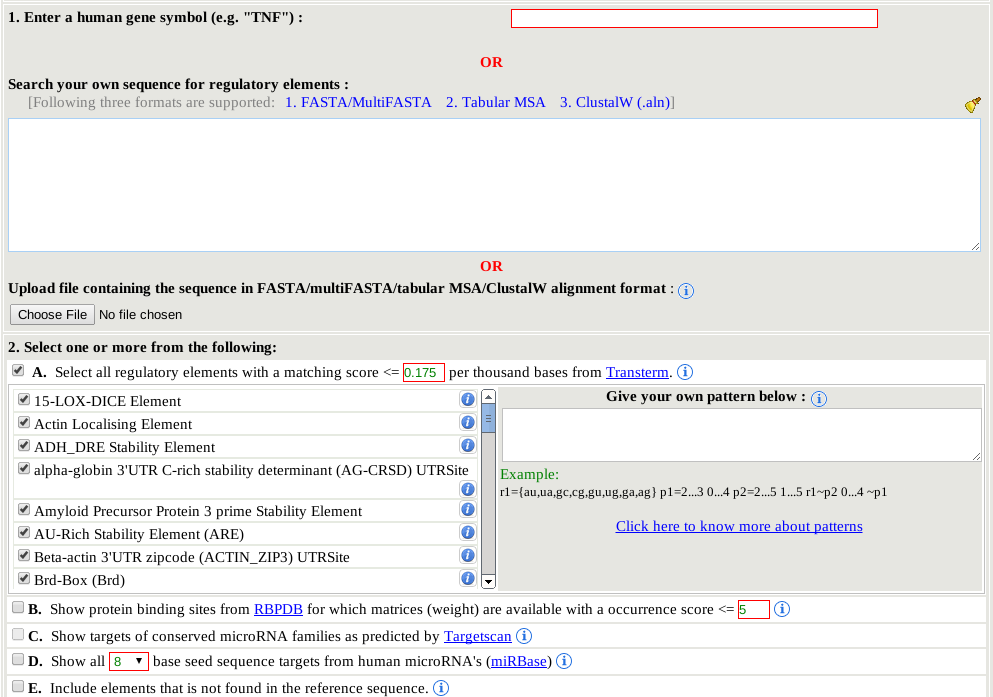
For all the elements provided by the Transterm database, their corresponding sequence pattern has been searched with Patsearch against dinucleotide shuffled sequences. The input sequence was 18,895 human UTRs from UCSC, made non-redundant. The average length was 1,281. The expected dinucleotide frequencies would be 0.0625 based on an equal amount of each base, however the dinucleotide frequencies were:
AA 0.088 AC 0.048 AG 0.068 AT 0.070 CA 0.068 CC 0.060 CG 0.012 CT 0.073 GA 0.057 GC 0.047 GG 0.057 GT 0.054 TA 0.061 TC 0.058 TG 0.078 TT 0.103TT and AA were the most overrepresented (0.103,0.088), CG most underpresented (0.012). The average number of matches per 1000 bases was recorded as the expected number of hit that would occur by chance (E-value) in shuffled sequences. A E-value per thousand is used as this is the typical length of a human UTR when SFM is run in the default mode, requiring a match to the human reference sequence to show a match in the output. As some of these elements have quite short sequences of identity (e.g. 8 nt), there is a high probability of false positive identification. An 8 base exact match would be expected in a UTR with equal amounts of the four nucleotides to occur once in every 4096 bases, therefore about 0.25 times in a 1000 bases 3' UTR. The actual E-values in dinucleotide shuffled sequences are higher or lower as some dinucleotides (e.g. AA, TT) are more frequent. The E-value filter can be changed by modifying the value ( default: 0.175) in the corresponding text box with red border. By changing the E-value in the textbox (a smaller value increases stringency), all the elements will be checked to see if their E- value per thousand bases falls above the specified value. Only the elements that have E-values below or equal to the specified value will be automatically selected. In section 2.A, every listed element has a small information icon at the end. By clicking on the icon, the user can refer to the complete details of the element without leaving the page (as shown in the right side image). This could be used to input data from other experiments, e.g. those in RBPDB, or from other experimental studies, or simply to visualise a sequence of interest. The user can also provide a custom sequence motif/pattern (e.g. 'GACTTT', or 'GAC 1....6 TT', or 'r1={au,ua,gc,cg,gu,ug,ga,ag} p1=2...3 0...4 p2=2...5 1...5 r1~p2 0...4 ~p1' or ^GTTGTT[1,0,0] ) to be included in the analysis. For a complete set of syntax rules refer to the original PatSearch paper (Grillo et al 2003) The user motif/pattern will be used along with the Transterm motifs (if any are selected) using PatSearch. In this case no E-value filer is applied. When the user is determining the biological significance of the result they should take into account the likelihood of a background match This is approximately 1/4096 for 6 nt of exact match (e.g. GACGTT, or GANNNCGNNTT), 1/16384 for 7 nt, and 1/65536 for 8 nt. Cite: Jacobs, G.H., Chen, A., Stevens, S.G., Stockwell, P.A., Black, M.A., Tate, W.P. and Brown, C.M. (2009) Transterm: a database to aid the analysis of regulatory sequences in mRNAs, Nucleic acids research, 37, D72-76. |
|
|
All 73 motifs from the RNA Binding Protein DB with PFM are included in SFM. The PFMs are searched against user input sequence using the command line version of MotifLocator (v3.2.0). Some of these PFM are short ~6-11 bases. The PFMs were searched against 10 randomly generated thousand base sequences and the occurrence per thousand bases was recorded. These E-values ranged from 0 to 44.
By modifying the E-value in the corresponding text box (a smaller value increases stringency) the user can omit matrices with high E-values. The default E-value cut off of 1 enables a search with approximately half the matrices.
It is also possible to use other binding sites from RBPDB by inputting the binding site sequences (e.g from the list of human RNA binding proteins here).
An example would be the reported binding sites for YBX1. There are twelve reported sequences from different studies using different techniques (e.g. UCCAGCAA link).
Version: v1.3 release 28.09.2012
Cite: Cook, K.B., Kazan, H., Zuberi, K., Morris, Q. and Hughes, T.R. (2011) RBPDB: a database of RNA-binding specificities, Nucleic acids research, 39, D301-308. |
 |
|
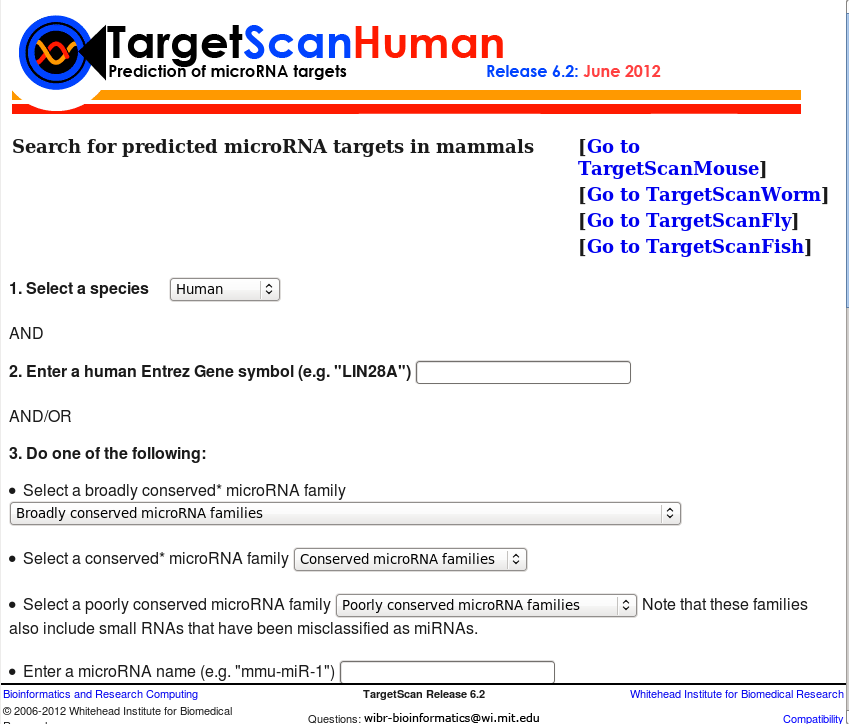
Targetscan is one of the most used and cited public database for microRNA binding site prediction and visualization, mainly focusing on orthologous 3’ UTRs of vertebrate sequences. It contains over 400 highly conserved targets of microRNA binding sites. All the "broadly conserved" and "conserved" microRNAs predicted target information (binding sites) and other associated files were obtained from the Targetscan website, processed and converted in the relational database table and added to Scan for Motifs.
Version: Release 6.2 (June 2012)
Cite: Garcia, D.M., Baek, D., Shin, C., Bell, G.W., Grimson, A. and Bartel, D.P. (2011) Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs, Nature structural & molecular biology, 18, 1139-1146. |
 |
|
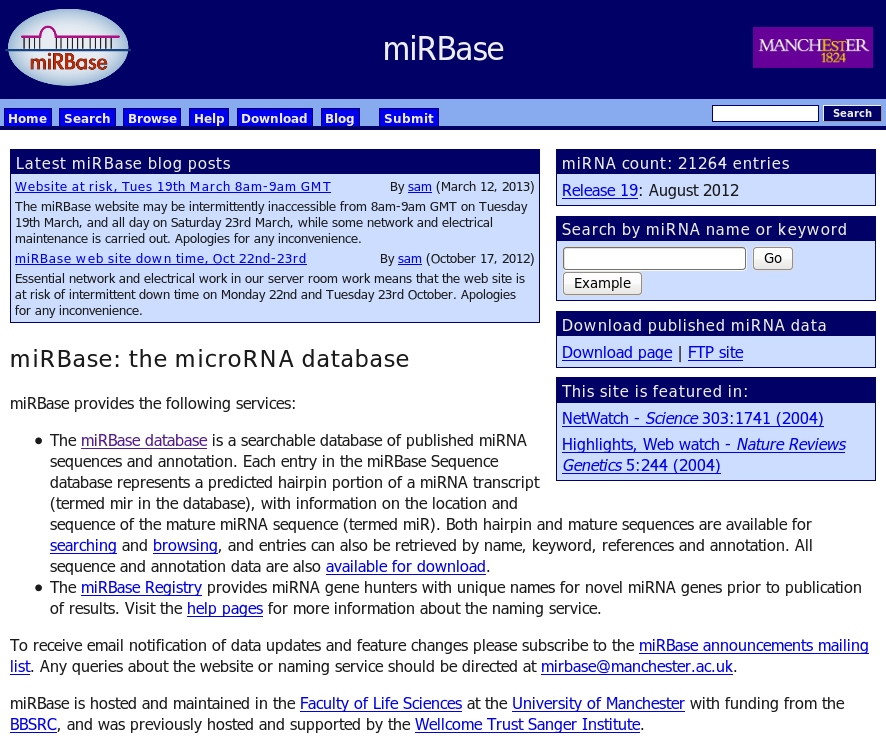
To visualize all the potential targets of miRNA binding sites in user input sequence, we downloaded the mature miRNA sequence file (mature.fa) from miRBase website, processed the file (reverse complemented and extracted 8 5' bases) to get a list of 2042 eight base long seed sequences, stored in a reference table and added to Scan for motifs server. These motifs are searched in the user sequence using perl's regular expressions.
The user can choose to show either 6 or 7 or 8 base long binding site targets by changing the corresponding value from the drop down list, controlling the number of motifs to be identified and shown in the output. It is expected that there would be approximately 1/4096 for 6mer of exact match, 1/16384 for each of the two 7mers, and 1/65536 for 8mer. As there are ~2000 seeds to be searched with, matches to short (e.g. 6mer) seeds must be interpreted with caution. There would be about 30 matches in total per 1000 base UTR from 7mer seeds (Bonferroni correction for multiple testing). In order to avoid false positives only Watson-Crick, A-U and G-C, not G-U base pairing is allowed.
To distinguish the two 7mers, the notation A1 is used for the 7mer.A1 (base 1-7). On visualising these results the adjacent features searched bor by miRNA target prediction programs could be used. For example, TargetScan would use: conservation across species, position in the UTR and local composition (Garcia et al 2011).
Version: Release 20 (June 2013)
Cite: Kozomara, A. and Griffiths-Jones, S. (2011) miRBase: integrating microRNA annotation and deep-sequencing data, Nucleic acids research, 39, D152-157. |
 |
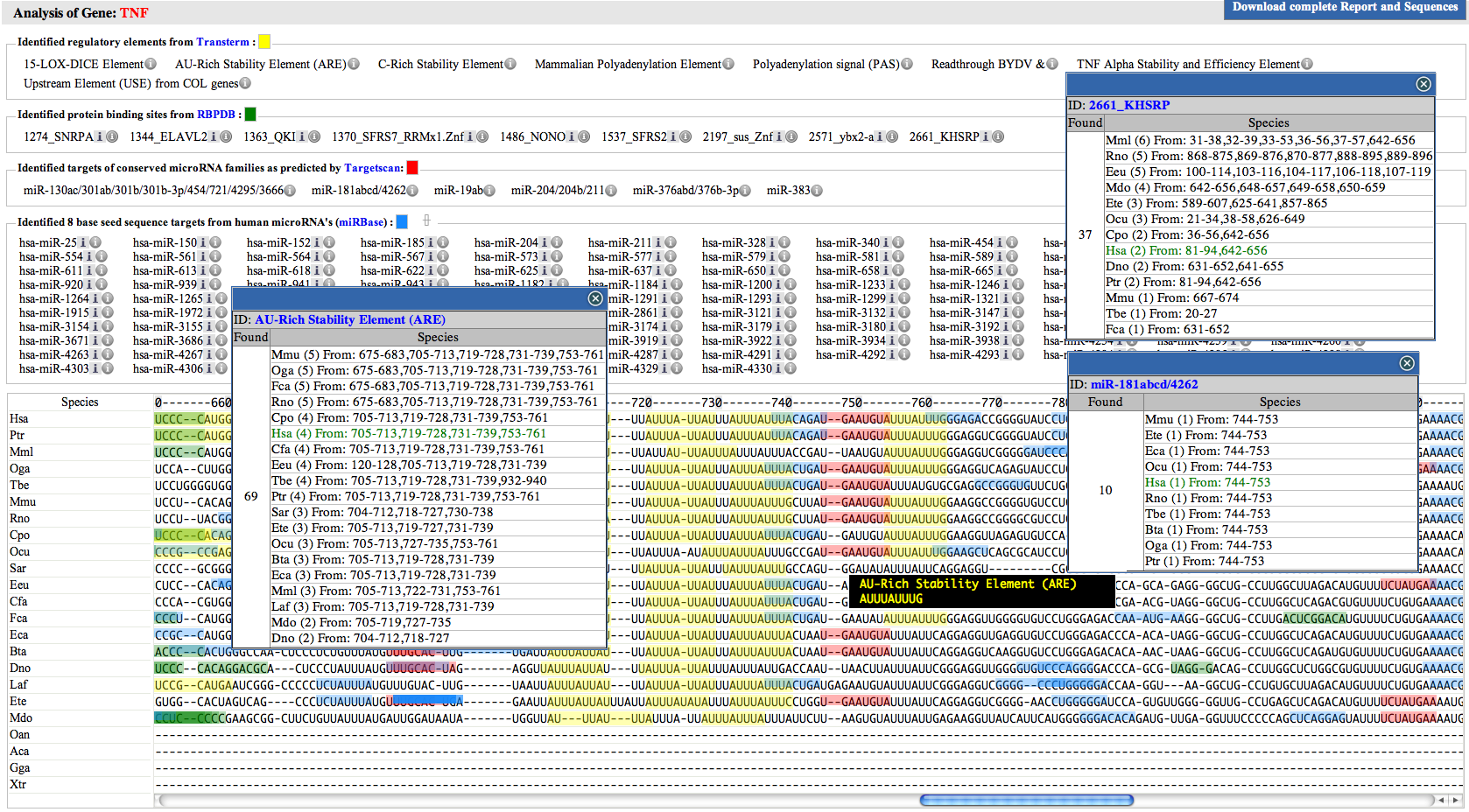
| The image on the right shows a typical "Scan for motif" output. Each type of regulatory element is grouped based on their source, and the identified elements are listed as links in the corresponding sections. The small icons on the right of these elements are linked to their sources. The square color boxes represent the colored codes of the corresponding elements. This means, every Transterm element has been highlighted with yellow color in the sequence. This highlight is partially transparent, allowing the user the ability to see all the other overlapping elements. The height of this colored boxes have different height, and presented in such layered way, that the user can mouse over on each element. Each identified element is linked to the first occurrene of that element in the sequence. Clicking on the element takes the current view point to the section. Because the miRBase seed sequences are only 6-8 bases long, they are the most frequently occuring elements in any general analysis (except FDR rate set to 3 or 3.5), The identified miRBase element section often contains so many identified elements, that user needs to scroll to see elements on the top list and their corresponding position in the sequence. To avoid this, the user may click on the plus/minus icon next to the "Blue square" on the right of "Identified 8 base seed sequence targets from human microRNA's (miRBase) :". This will toggle the view of miRBase elements. The bottom section shows the input sequence(s), highlighting every identified element. Placing the mouse on these colored boxes shows the element name and the motif sequence.By clicking on these colored boxes the user can see how many times and where in the input sequence this element was found. Finally, a detailed analysis report can be downloaded by clicking on the blue button named "Download complete Report and Sequences", which can very useful, particularly for two reasons. One, it has all the input parameters and sequences, which will allow the user to get all the experiment details in the future. Two, the identified elements are presented in "BED" format, allowing user to export the relative section(s) into other formats. A sample report can be obtained from this Link. | 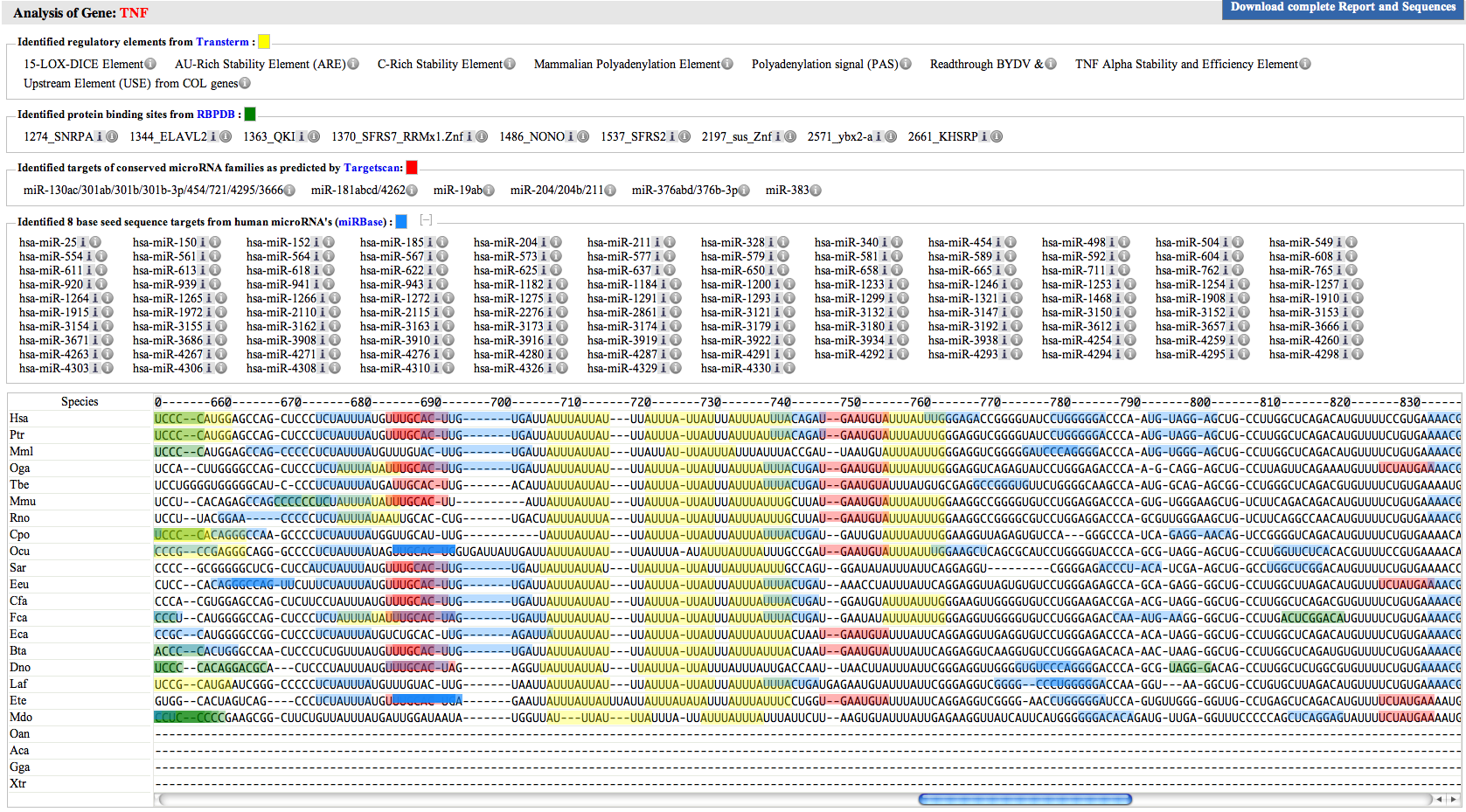 |
Places to start, reviews
Stevens, S. and C. Brown (2014). Bioinformatic methods to discover cis-regulatory elements in mRNAs. Springer Handbook of Bio-/Neuro-informatics. N. Kasabov. Heidelberg, Springer: 151-169.
Quattrone, A. and E. Dassi (2016) Introduction to Bioinformatics Resources for Post-transcriptional Regulation of Gene Expression (2016) Methods Mol Biol 1358: 3-28.
Specific Resources
Giudice, G., F. Sanchez-Cabo, C. Torroja and E. Lara-Pezz ATtRACT-a database of RNA-binding proteins and associated motifs (2016) Database (Oxford) April 2016.
Chang, T.H., Huang, H.Y., Hsu, J.B., Weng, S.L., Horng, J.T. and Huang, H.D. (2013) An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs, BMC bioinformatics, 14 Suppl 2, S4.
Claeys, M., Storms, V., Sun, H., Michoel, T. and Marchal, K. (2012) MotifSuite: workflow for probabilistic motif detection and assessment, Bioinformatics, 28, 1931-1932.
Cook, K.B., Kazan, H., Zuberi, K., Morris, Q. and Hughes, T.R. (2011) RBPDB: a database of RNA-binding specificities, Nucleic acids research, 39, D301-308.
Dassi, E., Malossini, A., Re, A., Mazza, T., Tebaldi, T., Caputi, L. and Quattrone, A. (2012) AURA: Atlas of UTR Regulatory Activity, Bioinformatics, 28, 142-144.
Garcia, D.M., Baek, D., Shin, C., Bell, G.W., Grimson, A. and Bartel, D.P. (2011) Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs, Nature structural & molecular biology, 18, 1139-1146.
Griffiths-Jones, S., Grocock, R.J., van Dongen, S., Bateman, A. and Enright, A.J. (2006) miRBase: microRNA sequences, targets and gene nomenclature, Nucleic acids research, 34, D140-144.
Grillo, G., Licciulli, F., Liuni, S., Sbisa, E. and Pesole, G. (2003) PatSearch: A program for the detection of patterns and structural motifs in nucleotide sequences, Nucleic acids research, 31, 3608-3612.
Grillo, G., et al. (2010) UTRdb and UTRsite (RELEASE 2010): a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs, Nucleic acids research, 38, D75-80.
Gruber, A.R., Fallmann, J., Kratochvill, F., Kovarik, P. and Hofacker, I.L. (2011) AREsite: a database for the comprehensive investigation of AU-rich elements, Nucleic acids research, 39, D66-69.
Jacobs, G.H., Chen, A., Stevens, S.G., Stockwell, P.A., Black, M.A., Tate, W.P. and Brown, C.M. (2009) Transterm: a database to aid the analysis of regulatory sequences in mRNAs, Nucleic acids research, 37, D72-76.
Kozomara, A. and Griffiths-Jones, S. (2011) miRBase: integrating microRNA annotation and deep-sequencing data, Nucleic acids research, 39, D152-157.
Lewis, B.P., Burge, C.B. and Bartel, D.P. (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets, Cell, 120, 15-20.